

From its’ inception in 2000, I led the establishment of the FDA/CDRH Human Factors program and then saw it flourish in its efforts to improve the safety and efficacy of medical devices. That initiative was then extended to FDA/CDER, as they began their Human Factors initiative for combination products. User Interface (UI) quality for medical products improved, as the review process matured. I witnessed numerous examples of user interface design flaws that caused problems as noted in formative tests (or “failed” summative tests) which subsequently were modified and corrected, resulting in a final version that was free from design-based use error, and risk to users and patients.
Industry dialogue was a key factor in growing the FDA program. Some manufacturers were bewildered at first about Human Factors, but after our discussions, they accomplished excellent work and produced first class products that were both safer and more effective when used. It was also gratifying when these manufacturers would call just to thank me for the help and offer their sincere appreciation for what the HF process did for their product and design process. Often, they employed test providers although some did excellent work with their own internal staff.
When I co-authored the current guidance for the FDA for Human Factors submissions for Medical Device, my intention was very clear. I wanted to develop a process that would;
• ENSURE THAT TESTING WAS A VALID AND AN EFFECTIVE SCREENER OF UI DESIGN FLAWS.
• ENSURE THAT DETECTION OF UI DESIGN FLAWS WOULD BE FOLLOWED BY CONSIDERATION FROM THE PERSPECTIVE OF THE USER, AS WELL AS PERFORMANCE AND OBSERVATION DATA TO ENABLE THE USE ERROR SCENARIO TO BE DESCRIBABLE.
• THAT UI DESIGN WILL SUPPORT INTENDED USERS, INTENDED USES AND, EXPECTED
USE ENVIRONMENTS. (SUPPORTS SAFE AND EFFECTIVE USE)
• PRESENT TESTING TO BE DONE IN A WAY THAT ALLOWS RESULTS TO BE GENERALIZABLE TO ACTUAL USE.
• PRODUCE SUFFICIENT AND APPROPRIATE DATA RELATED TO THE CONCLUSION OF SAFE AND EFFECTIVE USE.
• HF PROCESS WILL BE EASY TO FOLLOW AND EXECUTE.
• REQUIRE THE LEAST “BURDEN” TO INDUSTRY TO GET THIS DONE.
• SUPPORT EFFICIENT AND TIMELY REVIEWS BY FDA.
• ENSURE THAT TESTING WAS REPRESENTATIVE OF REAL WORLD USE.
Being “lean” is nice, but it is also a double-edged sword. Yes, you don’t always need to do a ton of testing and involve huge numbers of test participants, in fact you rarely do. The testing just needs to be robust, defensible, and generalizable to the real world of actual use.
The other side of the “lean” sword is this; if something important is omitted from this lean menu, the validation is incomplete. Likewise, if a critical piece is completed, but not done well (e.g. failing to collect post-test interview data), evidence of UI design adequacy would be missing, which the reviewer is looking for, so the review halts. Questions are asked. Time is spent, perhaps additional testing is required that could have as easily been carried out at the first attempt. I always have and continue to dislike it when that happens.
For example, absence of experiential data gathered from test participants, using the device overall, or the lack of analysis of test data to identify the source(s) of difficulty regarding critical task failures (assuming such are found) would present a mystery to the reviewer. FDA reviewers don’t like mysteries! I never did. What can you say in a review when data and analyses are missing?
FACED WITH MYSTERIOUS HF WORK, I WOULD WRITE A DEFICIENCY LETTER FASTER THAN
YOU CAN SAY “TEST PARTICIPANTS MADE ERRORS AND I DON’T KNOW WHY.”
When reviewing these aspects, (we reviewers must think about all those future users out there waiting to benefit from using the device.) How will they experience it? Will it save lives or will the user watch life fade before their very eyes while they try in vain to re-start the device for the 4 th time? This is a scenario no-one wants!
A flawed, or inadequate UI design, may well LOOK PRETTY IMPRESSIVE, AND A TEST REPORT BRIMMING WITH MULTIPLE FORMATIVE STUDIES MIGHT APPEAR TO BE COMPREHENSIVE, but a reviewer will and should not accept an incomplete, weak, or off-the-mark assessment.
REVIEWING ISN’T A “WHIFF” TEST, NOR IS IT A BOX-CHECKING EXERCISE.
AN HF REVIEWER, WORTHY OF THE POSITION, SHOULD NOT ACCEPT WEAK, INCOMPLETE, OR OTHERWISE DEFICIENT TESTING.
WHAT IS DEFICIENT, (AND MYSTERIOUS) HF WORK? GOOD QUESTION! I’M GLAD YOU ASKED!
Its work that doesn’t build a pyramid like the one shown here because one or more layers underneath the Conclusions (top) wasn’t completed or was done poorly (or, bizarrely). The higher layers only exist in a rational way when the layers below are solid.
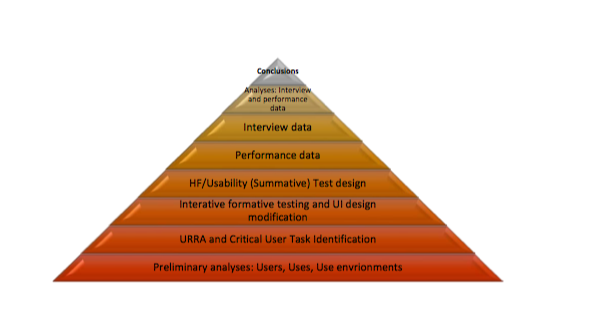
Indeed, there are good things to discuss about each of these layers. But, why might a HF effort fail to complete the pyramid? Why would they complete all the other work and botch a layer or two? That’s a shame. It’s a shame to waste effort and resources in this way.
IN MY EXPERIENCE OF BOTH REVIEWING AND CONSULTING, I THINK I UNDERSTAND SOME OF THE MAJOR REASONS WHY.
It’s not because someone didn’t attend a class and learn the techniques for how to slog through HF evaluation and testing, step by step, without understanding the underlying test purpose or theory.
It’s not because somebody didn’t get the “latest thinking from the FDA”. (BTW there isn’t a lot of this, other than a couple of Guidance notes by CDER, the fundamental and 98% of what you need to do is in this pyramid, and that isn’t changing any time soon.) Apart from an explicit discussion of Use Related Risk Analysis (URRA) which was not emphasized in the CDRH guidance, you can use it to do the tasks required and it’s almost easy!
But like the aviation analogy of “controlled flight into terrain” there are folks busily doing HF work that doesn’t build a pyramid once the layers are examined. (This is a fine condensation of what the HF review does – It looks through the pyramid layers.) The big question is “Can they can come to conclusions and perform the evaluation in a different and more efficient way?” Nope. It doesn’t happen, I’ve seen it all. They are hoping the reviewer will believe conclusions on faith with no structure (pyramid) beneath them.
SO WHY DOES THIS HAPPEN? A FAULTY PYRAMID, WITH SQUASHED MANGLED WRECK OF AN HF EFFORT, OR ONE WITH A HOLE SO BIG IT CAN’T SUPPORT THE LAYER ABOVE.
The answer has a lot of parts to it and I have a lot I can say about all of them. This paper is too short to discuss all of that, but to start you on this path, I’m going to introduce you to “locks” and “keys”.
Think of locks as ideas and (mis)information or “mindsets” that prevent those involved from building a good solid HF pyramid that will withstand scrutiny. “Keys” are information that open the locks. ”Keys” make you say “Oh, I see why. I see how, that’s easier than I realized!”
Currently, I see five major kinds of “locks”:
1. LEAVING OUT A LAYER – This is obvious, a pyramid can’t be built on thin air. The structure will crumble, as I have mentioned. No brainer.
2. MISUNDERSTANDING WHAT EFFECTIVE HF WORK IS – If you have belief that “expert review” adequately predicts, as well or better, than well executed simulated use testing with representative users, you should stop and take a breath. You currently believe one thing that is not correct. There might be others, be careful! The FDA wants you to have rated your use-related hazards via Risk Priority Numbers (RPN). Oh my, be careful!
3. NOT UNDERSTANDING WHY ANY TINY BIT OF HF WORK IS NECESSARY, OR WHY IT IS
IMPORTANT TO FDA REVIEW – Yes, the “Why”. There is always a reason why that makes sense. If it doesn’t make sense, you need to keep learning until it does. If you think the reason for testing is anything other than refining and then validating the design of the user interface, you’re mistaken! There is no other reason to do HF.
4. BEING MISINFORMED – Do you really know why you need to test what you need to test, and with how many people and, equally importantly, do you know what you don’t need to do? Are you expert enough to challenge whoever has advised you to design the test like this? You need to be. People are always being sold bags of goods that sound easy and glamorous. Don’t believe it. The pyramid’s glamor is that it works and works efficiently.
5. NOT UNDERSTANDING /MISINTERPRETING FDA FEEDBACK – The amount of misinterpretation, speculation and misunderstanding of what the FDA says in presubmission (Type C) meetings or in deficiency letters, is surprising and unfortunate. The same can be said of what the Agency says in public presentations. People walk away with the wrong ideas about what was just said. This doesn’t need to be the case. This can set up a stealthy chain of misfires that can ruin an entire effort.
6. NUMERICAL VALUES, RATING SCALES, STATISTICAL POWERING, RPN – If you think you can apply statistics to this stuff, you not only don’t understand this process, you don’t understand statistical theory either. But, there’s too much on this quantitative vs. qualitative discussion to cover much in this paper.
ABOUT RON KAYE
Ron Kaye recently retired from the FDA’s Center for Devices and Radiological Health where he led the development of its Human Factors initiative during his 19year tenure at the agency. Ron was the lead author of the original FDA human factors guidance released in 2000, and the current HF guidance released in February 2016, which represents the perspective of the FDA on pre-market submission human factors requirements. During his time at CDRH, Ron participated in over 1000 new device reviews involving human factors work submitted to almost all CDRH divisions, has trained FDA inspectors in HF, and has participated in Agency post-market responses and recalls associated with use error issues.
Ron has been integrally involved in the education of the FDA and device manufacturers regarding the human factors process in device design and testing. Ron’s participation as a faculty member of the AAMI HFE short course has brought the FDA human factors message to over 1200 industry practitioners over the past seven years, resulting in a significant improvement in human factors work for new device submissions. He has also been a coauthor of the AAMI/ANSI (HE-75) Standard, Human Factors in the Design of Medical Devices and has been a member of the international working group that produced IEC 62366, Application of Usability Engineering to Medical Devices.
DO YOU WANT TO LEARN MORE?
There is only one course taking place, anywhere in the world in 2018, where you will receive direct tuition and guidance from Ron and his colleague, Dr. Robert North.
They will show you how to build a robust and easy to understand pyramid and how to ensure you do not misunderstand direction from the FDA and anyone else outside your organisation. In addition, Ron and Bob will expand in detail on all the “locks” and “keys” introduced in this white paper.
This course takes place in Cambridge, UK from 13th to 15th November 2018 and places are strictly limited.
If you would like more information, please
CLICK HERE
to view our one-page brochure.
If you would like to book a place, please get in touch with Sean Warren at The Moon on a Stick at
sean@the-moon-on-a-stick.com